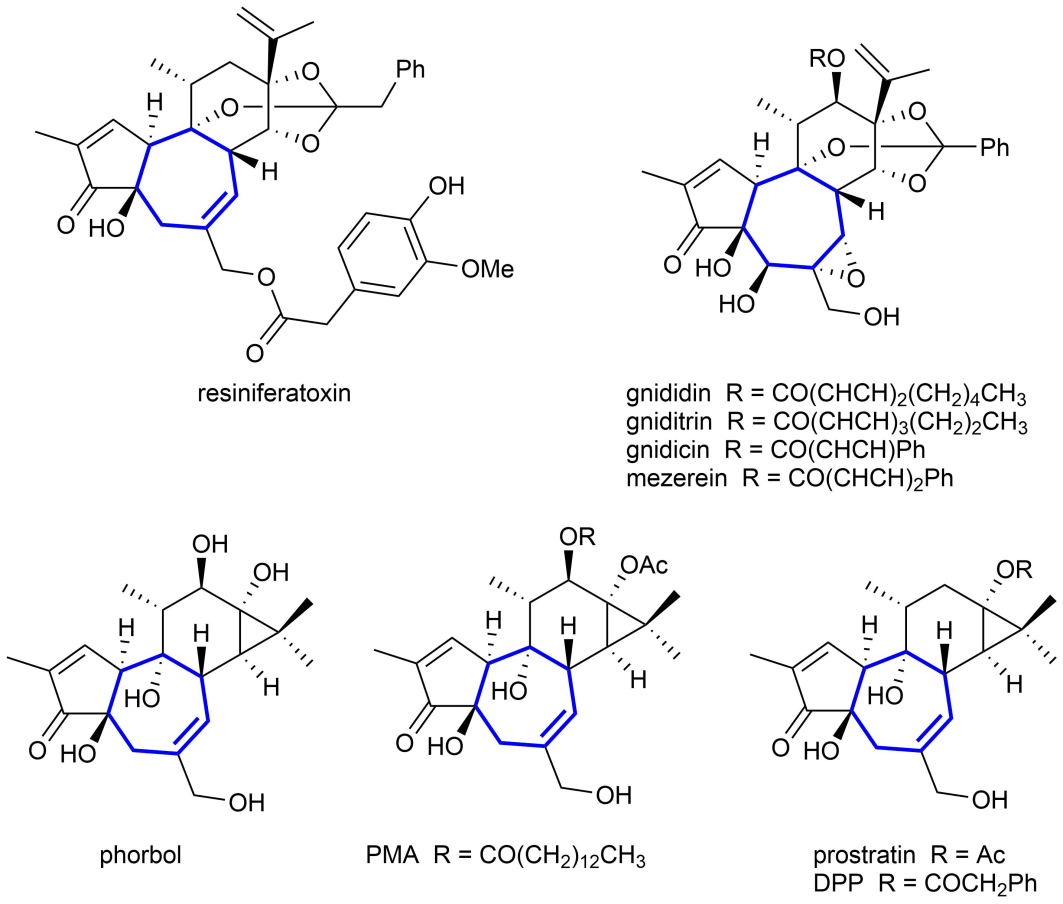
受到萜类化合物生源合成途径的启发,有机化学家设计并发展出了许多高效的多烯串联环化反应。虽然这些反应可被用于合成含有五元和六元碳环的天然产物,但是由于环化过程中存在着不利的跨环相互作用和熵效应,此类反应往往很难被用于七元碳环等中环体系的构筑。图1中展示了一些含有七元碳环结构的天然产物。
图1. 一些含有七元碳环结构的天然产物。
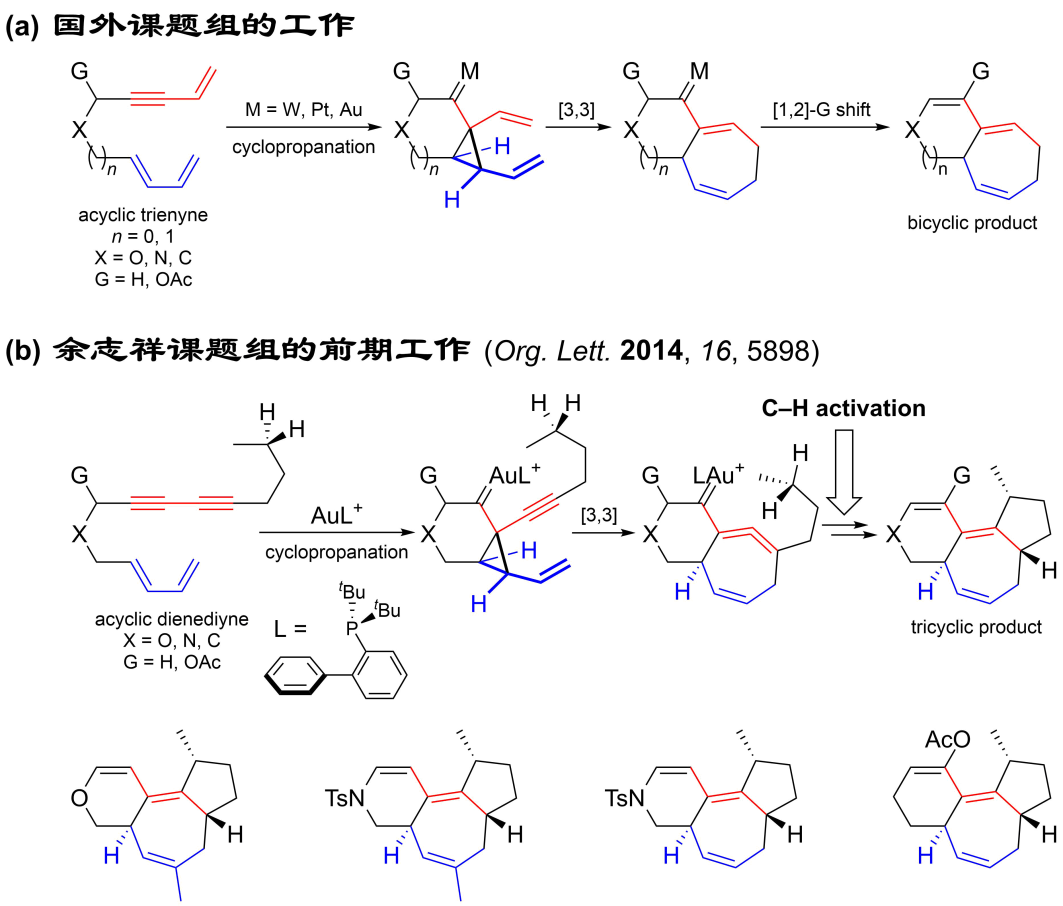
1,2-二乙烯基环丙烷的Cope重排是一种合成七元碳环的方法,但是底物的合成通常较为繁琐。为了解决这一问题,合成化学家后来发展出了过渡金属催化的串联环丙烷化/Cope重排反应,实现了6-7和5-7双环体系的高效构筑(图2a)。受到这些工作的启发,合成与组装化学研究部余志祥课题组于2014年发展了一种金催化的双烯双炔环化异构化反应(该反应亦可被称为形式[4+3]环加成/碳氢键活化反应),通过巧妙地引入1-乙烯基-2-炔基环丙烷的Cope重排反应,高非对映选择性地合成了具有七元碳环的6-7-5三环化合物(图2b)。作者利用金催化的串联环丙烷化/Cope重排/碳氢键活化反应,依次构建了三根碳碳键和三个环,从而实现了由简单线性分子到复杂多环分子的高效转化。此外,通常情况下惰性烷基碳氢键活化需要相对苛刻的反应条件,而在此工作中碳氢键活化反应在室温下即可顺利发生。
图2. (a)串联环丙烷化/Cope重排反应;(b)串联环丙烷化/Cope重排/碳氢键活化反应。
最近,余志祥课题组又对该反应的机理进行了研究。密度泛函理论计算和氘代实验表明该反应经由配体交换、环丙烷化、1-乙烯基-2-炔基环丙烷的Cope重排、碳氢插入和[1,2]-氢迁移等基元步骤完成(图3)。其中,反应的决速步是环丙烷化步骤。通常情况下,通过1-乙烯基-2-炔基环丙烷的Cope重排得到的七元环联烯在合成上的价值不大,这是由于其可以发生后续的二聚反应。而在余志祥课题组的工作中,七元环联烯C具有一个烯基正离子共振式D。烷基碳氢键的活化正是通过该烯基正离子对碳氢键进行快速插入予以实现的(理论计算结果表明这一步的活化Gibbs能仅为1.5 kcal/mol)。
图3. 催化循环。
上述反应和机理适用于X为O或NTs的底物(当X为CH2且迁移基团G为OAc时,机理有相应的变化),而不适用于双酯桥底物(当X为C(CO2Me)2且迁移基团G为H时)。这是由于氧桥和氮桥底物的环丙烷化具有endo-dig选择性(图4a,左),从而能够引发后续的Cope重排;而双酯桥底物的环丙烷化具有exo-dig选择性(图4a,右),不能引发后续的Cope重排。基于此,作者提出了一个设想:如果使用缩碳底物,则exo-dig环化将生成具有高度张力的4-3并环骨架,从而得到抑制。作者进一步的密度泛函理论计算支持了这一观点:相对endo-dig环化而言,exo-dig环化在动力学上较为不利。基于这些理论预测,作者随后在实验上成功地实现了缩碳底物的环化异构化反应,为构建具有全碳骨架结构的5-7-5三环和5-7-6-6四环化合物提供了新的合成工具(图4b)。在这个新的环化异构化反应中,2a的合成仍然通过烷基碳氢键活化实现,而在2b-d的形成过程中则改用了Friedel-Crafts反应。这一新反应的实现很好地展示了余志祥课题组理论与实验相结合的研究理念在解决合成化学实际问题中的优势。
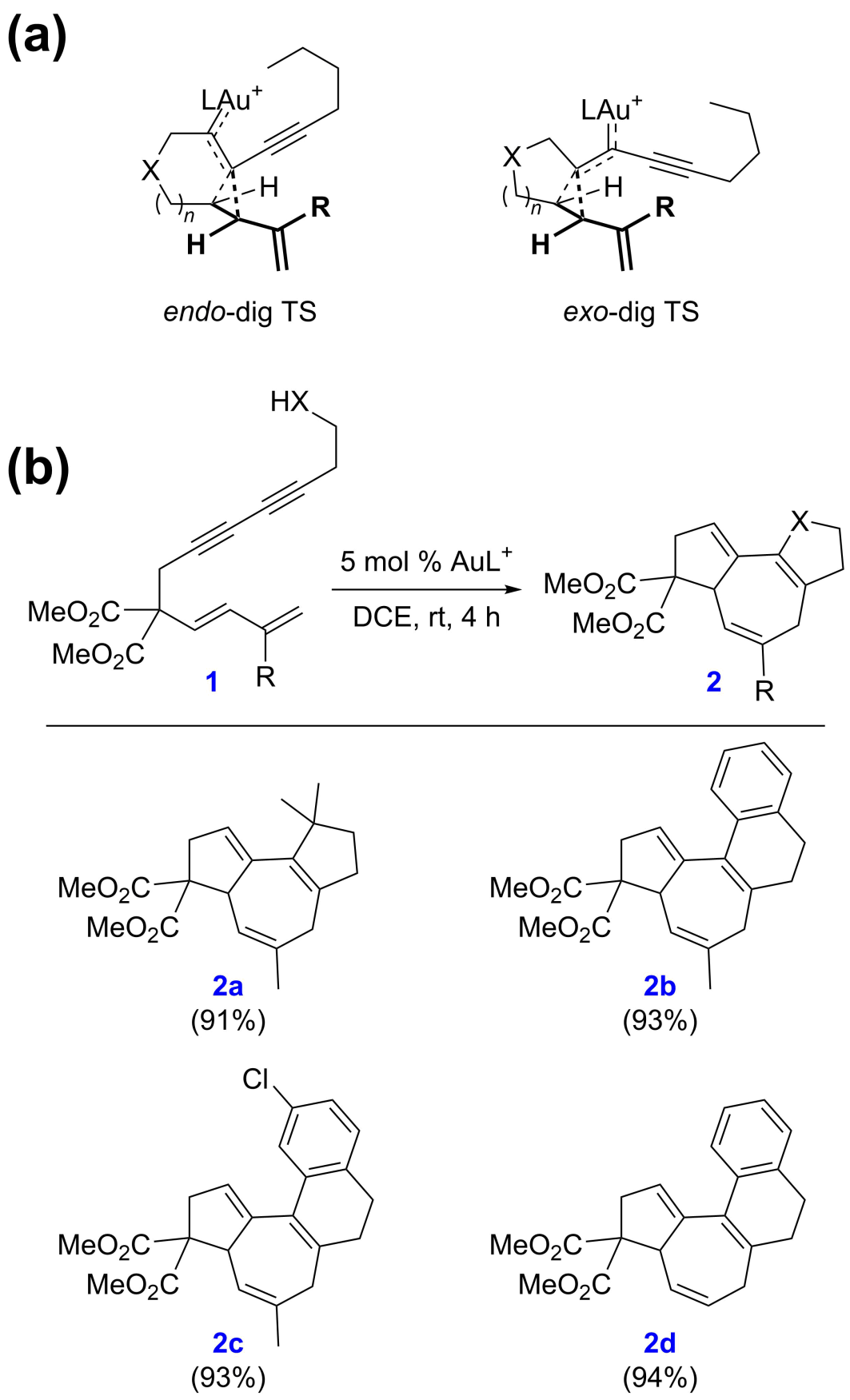
图4. 通过机理研究设计新的环化异构化反应。
该课题的前期工作(合成方法学部分)由余志祥课题组的蔡沛君博士、王熠博士和柳成航博士完成,发表在Org. Lett. 2014, 16, 5898上。最近的机理研究工作由王熠博士和蔡沛君博士完成,发表在J. Am. Chem. Soc. 2020, 142, 2777上。