陈辉课题组在理论计算研究铁促发[2+2],[2+1]烯烃环加成反应机理方面取得突破
Diels-Alder[4+2]环加成反应,是应用极其广泛的六元碳环合成方法。而对于更小的四元碳环的合成,根据Woodward-Hoffmann分子轨道对称守恒原理,协同的[2+2]环加成反应在基态时轨道对称性不匹配,将产生显著的动力学能垒,阻碍反应的发生。因此,相比于较成熟的[4+2]环加成反应,如何促使烯烃[2+2]环加成反应的进行,是有机合成方法学研究中的前沿领域之一。协同的烯烃[2+2]环加成反应涉及两根C-C键的协同形成,实验研究发现,利用一些廉价过渡金属中心,可将该协同过程分解为一次只形成一根C-C键的分步[2+2]环加成,该过程可有效降低[2+2]环加成反应的能垒,但其背后的机理,特别是开壳层廉价金属各自旋态、以及其氧化还原配体所起的作用,都还不清楚。这些问题亟待理论计算化学研究去解决。
在中科院化学所理论计算化学平台项目和国家自然科学基金委的支持下,化学所光化学院重点实验室的科研人员在理论计算研究铁促发[2+2]及[2+1]烯烃环加成反应机理方面取得突破。在前期工作中,科研人员与中科院上海有机所相关课题组合作,对三齿膦氮配位环境的铁叶立德化合物的电子结构、及其与烯烃的[2+1]环加成反应的机理开展了密度泛函理论计算研究。研究发现,铁叶立德化合物在与烯烃的反应中,可异构化为不稳定的铁卡宾中间体,从而与烯烃发生[2+1]环加成反应生成环丙烷。故而铁叶立德化合物可成为一种潜在的活性铁卡宾前体(图1)。相关结果发表在J. Am. Chem. Soc. 2017, 139, 3876-3888。
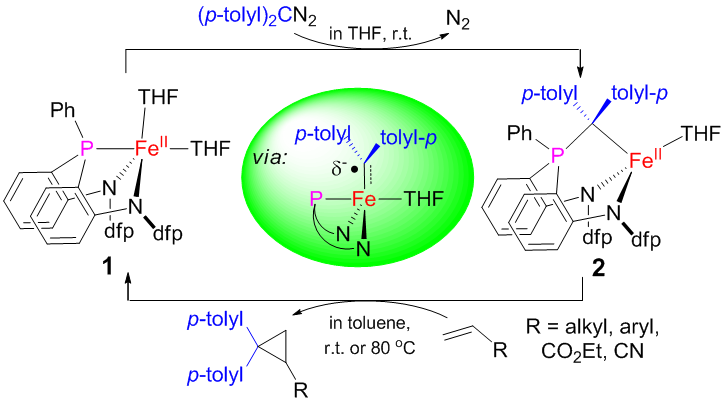
图1 理论计算揭示铁叶立德化合物(2)可通过转变为铁卡宾中间体完成与烯烃的[2+1]环加成反应生成环丙烷类产物
最近,针对实验化学家发现的铁催化[2+2]烯烃环加成反应,利用高精度多参考波函数方法(CASPT2)结合密度泛函方法,科研人员首次发现,铁催化[2+2]烯烃环加成反应具有底物依赖的二态反应性。研究发现:(1)单烯间的[2+2]环加成反应,与单烯/共轭二烯间的[2+2]环加成反应,其反应速度决定步骤C-C偶联过程具有不同的反应图像:前者具有单态反应性,而后者是二态反应性(图2);(2)氧化还原活性PDI配体在铁催化[2+2]环加成反应所起的作用,在于避免热力学上不利的Fe(II)/Fe(0)还原消去C-C偶联,产生热力学上更有利Fe(III)/Fe(I)还原消去C-C偶联,并且PDI配体在还原消去C-C偶联中作为电子受体的作用也是底物依赖的;(3)实验发现的单烯/共轭二烯间的交叉[2+2]环加成反应较低的反应性主要是由于:(a)单重态上不利的反应能,(b)单重态到三重态较低的自旋翻转效率。科研人员预言改善单重态到三重态的自旋态翻转效率将有利于提高交叉[2+2]环加成反应的反应性。该工作不仅揭示了铁催化C-C偶联反应的机理,也成功拓展了多参考波函数方法的应用范围。相关结果发表在J. Am. Chem. Soc. 2017, 139, 15564-15567。
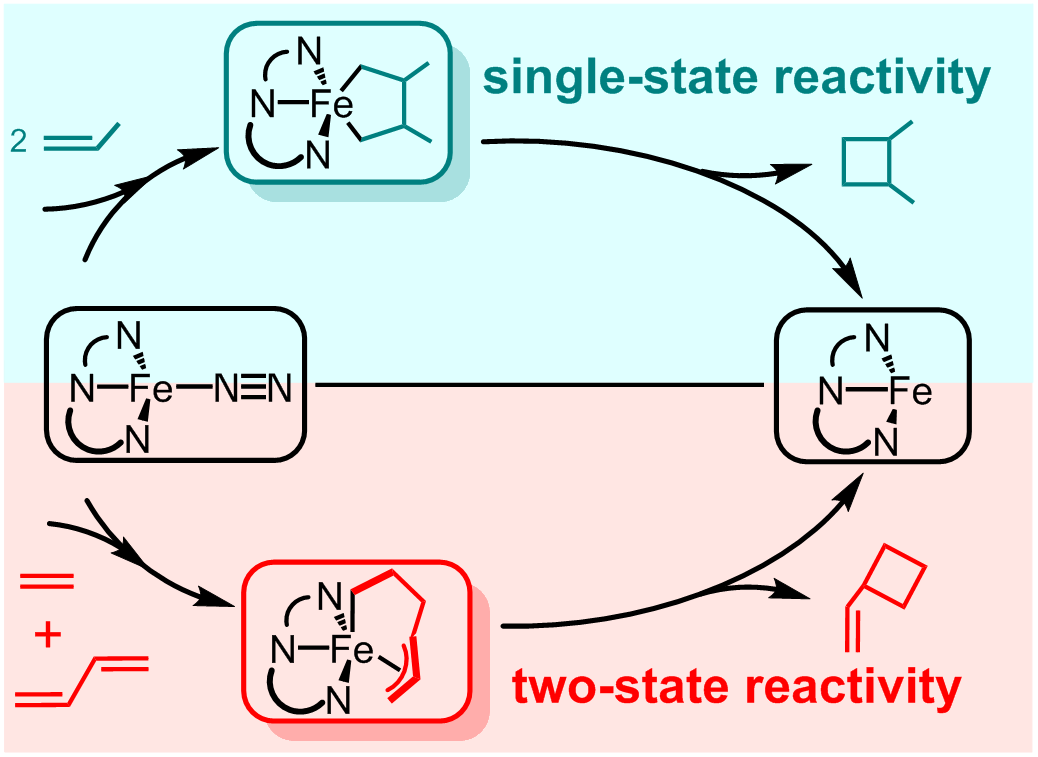
图2 CASPT2/DFT理论计算揭示铁催化[2+2]烯烃环加成反应机理的底物依赖性